
Our goal is to provide a comprehensive mechanistic understanding of functional genomics, gene regulation, and neural circuits, and to enhance genotype-phenotype predictions, particularly for human brains and brain diseases. To this end, we develop and apply machine learning and artificial intelligence (ML/AI) methods and bioinformatics tools that enable prioritizing multimodal features and feature relationships (e.g., genes to networks to circuits) for disease and clinical phenotypes. Our tools are open-source available with tutorials, workflows, and demos on our Github site.

Genotype-Phenotype Prediction and Functional Genomics. Genotypes are strongly associated with disease phenotypes, particularly in brain disorders. However, the molecular and cellular mechanisms behind this association remain elusive. To address this, we developed several deep learning methods including Varmole, ECMarker and DeepGAMI to improve genotype–phenotype prediction from multimodal data. They leverage functional genomic information, such as eQTLs and gene regulation, to guide neural network connections. Additionally, DeepGAMI includes an auxiliary learning layer for cross-modal imputation allowing the imputation of latent features of missing modalities and thus predicting phenotypes from a single modality. Finally, we use the integrated gradient to prioritize multimodal features for various phenotypes.
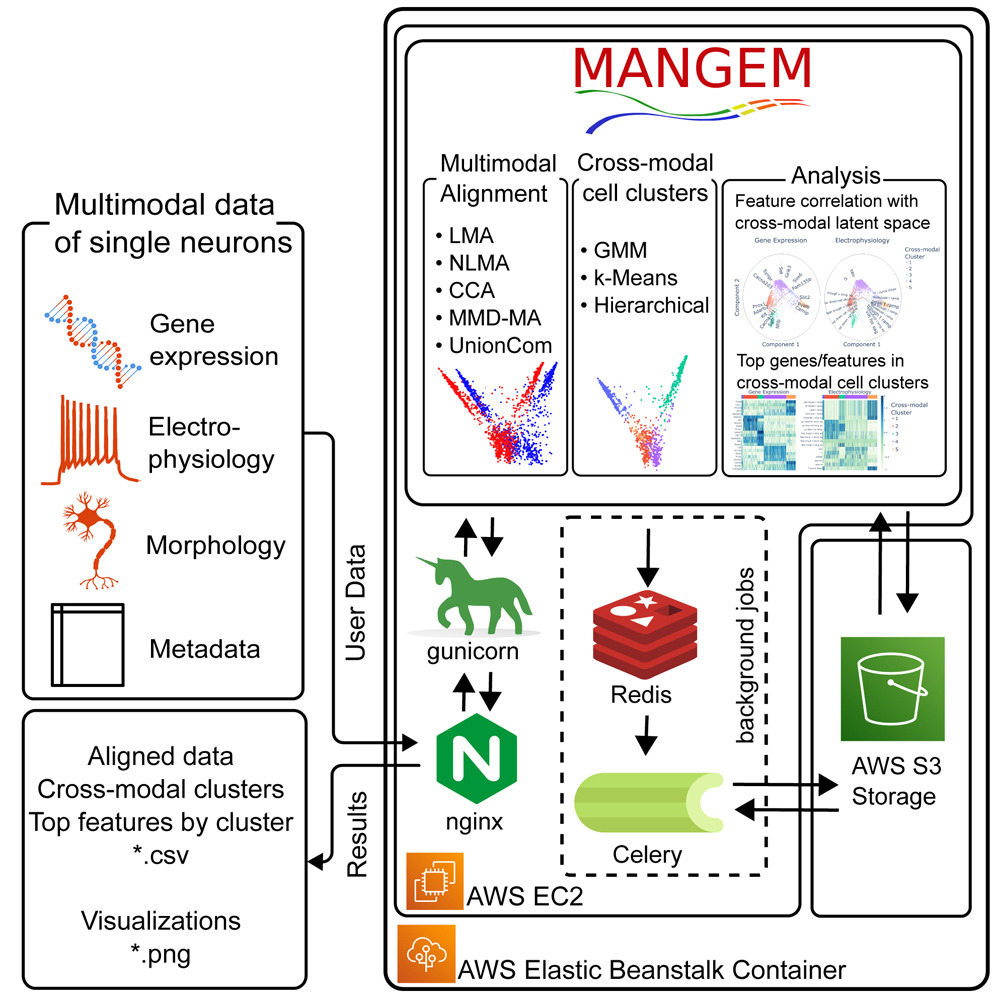
Single-cell Multimodal Learning. Multimodal measurements of single-cell sequencing technologies facilitate a comprehensive understanding of specific cellular and molecular mechanisms, including scRNA-seq, scATAC-seq, spatial organization, and electrophysiology. However, simultaneous profiling of multiple modalities of single cells is challenging, and data integration remains elusive due to missing modalities and cell–cell correspondences. To address this, we developed a framework called Multiview Empirical Risk Minimization (MV-ERM) unifying different multimodal learning approaches. Using MV-ERM, we recently developed several methods such as deepManReg, JAMIE, CMOT, MANGEM and COSIME for multimodal integration, cross-modality imputation, and phenotype prediction. Also, our methods can prioritize multimodal features such as genes, and open chromatin regions for imputation and prediction.

Dynamic Modeling and Analysis of Gene Expression and Regulation. To understand dynamic changes in gene expression and regulation in complex biological processes, we developed several machine-learning methods to identify represented gene expression patterns in development. For instance, our DREISS method used the state-space model to compare embryonic developmental gene expression data across model organisms and infer cross-species conserved and species-specific expression patterns representing rising, falling, and oscillating genes. Recently, we developed a manifold learning method, BOMA for comparative developmental gene expression analysis across conditions (e.g., brains and organoids) to identify conserved and specific developmental trajectories as well as developmentally expressed genes and functions, especially at cellular resolution.

Single-cell Network Biology and Medicine. Gene regulatory networks fundamentally control gene expression. We developed several computational methods (e.g., scGRNom) for predicting gene regulatory networks linking transcription factors, regulatory elements, and target genes, especially at the cell type level. Recently, we analyzed regulatory network structures and circuits such as hierarchy and motifs and then used the network structures to repurpose drugs at the cell-type level (e.g., co-regulation modules) for Alzheimer’s disease and neuropsychiatric disorders. Furthermore, we developed a novel manifold learning method, ManiNetCluster, simultaneously aligning and clustering gene networks to reveal the links of genomic functions between different conditions.
